
The purpose of this research
Methylated DNA-binding protein MeCP2, protein kinase CDKL5, and transcription factor FOXG1 are known as causative genes for Rett syndrome, but the relationship between these gene mutations and Rett syndrome pathology has not been clarified.
Recently, we identified a new CDKL5 mutation (Y177C). Therefore, the aim of this study is to elucidate the pathogenic mechanism of the CDKL5 mutation in Rett syndrome. Finally, we wish to produce a drug for treating Rett syndrome.
What is Rett syndrome

Rett syndrome is a rare disease that is associated with mental retardation, autism, stereotyped movements of the hands. MeCP2, CDKL5, and FOXG1 have been identified as causative genes for this disease.
 Details of Rett syndrome symptoms
Details of Rett syndrome symptoms
Symptoms

Symptoms of Rett syndrome caused by a mutations of MeCP2 gene include self-inflicted tendency and intellectual disability as seen from infancy. In addition, this disorder is characterized by its repetitive hand movements. Moreover, epileptic seizures, convulsions, and breathing abnormalities are often seen.
Diseases caused by mutations in the CDKL5 and FOXG1 genes are called atypical Rett syndrome. Patients with CDKL5 mutations are known to experience earlier epileptic seizures compared with MeCP2 mutations, and patients with FOXG1 mutation may develop microcephaly.
Pathogenesis
As mentioned above, it has been revealed that loss of function mutation in MeCP2, CDKL5, and FOXG1 genes is an etiology of Rett syndrome.
Treatment
Effective treatments and preventive methods have not been established to date and patients only receive symptomatic treatment.

Our challenge
MeCP2 is identified as a causative gene for Rett syndrome and its protein regulated epigenetic systems through binding into methylated DNA. Most patients with Rett syndrome have MeCP2 mutations, but some patients have CDKL5 or FOXG1 mutations, thereby causing atypical Rett syndrome.
We previously reported a novel CDKL5 mutation (Y177C) from a Japanese atypical Rett syndrome patient. Since this mutation site was observed within the catalytic domain, we considered that this mutation led to a loss of catalytic activity.
When we examined the enzymatic activity in vitro, we could not detect the expected CDKL5 (Y177C) activity. This finding was the first report to link gene mutation with loss of enzymatic activity in our research field.
However, since the relationship between the disappearance of activity of CDKL5 and the pathology of Rett syndrome has not been elucidated at all, we have attempted to clarify the mechanism of the CDKL5 mutation on the pathology at the molecular and cellular levels.
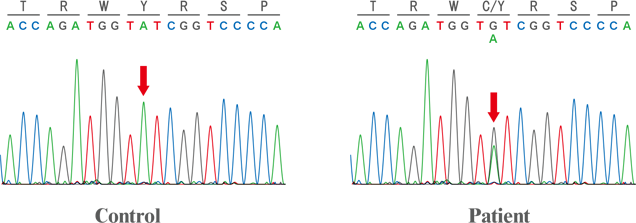
We analyzed the CDKL5 gene sequence for a patient with Rett syndrome and determined a new mutation site of CDKL5 (red arrow in the upper figure). This mutation was substitute tyrosine (Y)-177 to cysteine (C).
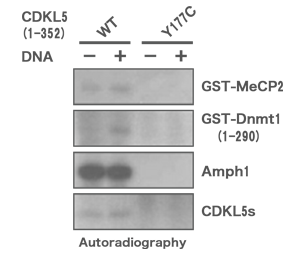
When we examined the enzymatic activity in vitro, we could not detect the expected CDKL5 (Y177C) activity.
Project members
Tetsuya InazuProfessor, College of Pharmaceutical Sciences, Department of Pharmacy
Masahiro ItoProfessor, College of Life Sciences, Department of Bioinformatics
Takako KawanoAssociate Professor, College of Pharmaceutical Sciences, Department of Pharmacy
Yukihiko KubotaAssistant Professor, College of Life Sciences, Department of Bioinformatics
Other Research subjects
Cause elucidation of transporter dysfunction in Fanconi syndrome and Bartter syndrome.
Fanconi syndrome (FS) is the general functional disorder of proximal tubules, and is a syndrome in which many substances like glucose, amino acids and phosphate cannot be reabsorbed and wasted in urine. Consequently, rickets is observed in child patients whereas osteomalacia is observed in adult patients. In this research program, we try to study the detailed development mechanism and preventation of Fanconi syndrome (FS).
In this research program, we try to study the detailed development mechanisms of Fanconi syndrome (FS) and Bartter syndrom (BS), and their preventation.
Functional analysis of the gene products related to Prader-Willi syndrome
Prader-Willi syndrome (PWS) is a neurodevelopmental disorder caused by loss of expression of paternal genes on chromosome 15. Major characteristics includes short stature, obesity, hypogonadism, respiratory distress, pain insensitivity and cognitive delays.
We will focus on the features of PWS that are related to obesity in order to understand the mechanisms involved in its pathogenesis and to explore novel therapeutic options.
Elucidation of the mechanism underlying laminopathies and development of their therapies
The goals of our research are to elucidate the mechanism underlying two laminopathies and to develop new therapies for these diseases by cutting-edge methods of various research fields including proteomics, cell biology, in silico structural biology, and drug design.
