
The purpose of this research
Our research goals are to elucidate the underlying mechanisms of two laminopathies and develop new therapies for these diseases using cutting-edge methods from various research fields, including proteomics, cell biology, in silico structural biology, and drug design.
What is the disease called laminopathy?
Genomic DNA is stored inside the nucleus and the expression of the genetic information is controlled by minute mechanisms involving various nuclear proteins.
Double membranes separate the nucleus from the cytoplasm. Nuclear lamina are on the nucleoplasmic side of the inner nuclear membrane; their main component is Lamin A, which provides a framework for the nuclear envelope.
Many proteins are located in the nuclear lamina and play important roles in the regulation of various nuclear functions. Diseases caused by mutations of the genes encoding these proteins are called laminopathies.
Our research aim is to elucidate the underlying mechanisms of laminopathies, including Néstor-Guillermo progeria syndrome (NGPS), Hutchinson-Gilford progeria syndrome (HGPS), and Emery-Dreifuss muscular dystrophy (EDMD), and to develop therapies for these rare diseases.
 Néstor-Guillermo progeria syndrome (NGPS)
Néstor-Guillermo progeria syndrome (NGPS)
One of the laminopathies is Néstor-Guillermo progeria syndrome (NGPS), a progeria syndrome, which is caused by the mutation of the BANF1 gene encoding the barrier-to-autointegration factor (BAF). The BAF is located into the inner nuclear membrane by interacting with the proteins bound to the nuclear lamina.
The BAF is reported to play important roles in the various gene expressions and the repair of DNA damaged by ultraviolet radiation. Thus, the decrease in DNA-binding activity of BAF induced by the mutation is thought to cause its hypofunction, leading to the onset of NGPS.

The estimated incidence of NGPS is one in approximately one million people. NGPS is characterized by growth retardation, dryness and atrophy of the skin, osteoporosis, and osteolysis during the childhood period of a patient. Symptoms such as diabetes, hypertriglyceridemia, and heart vascular disorder are not seen in the adulthood period, and the patients’ aging progresses slowly in comparison with HGPS. NGPS is defined as chronic progeria because the average life span of patients with NGPS is relatively long.
The details of the onset mechanism of NGPS have not yet been elucidated; therefore, no prophylaxis or therapy have been developed yet.
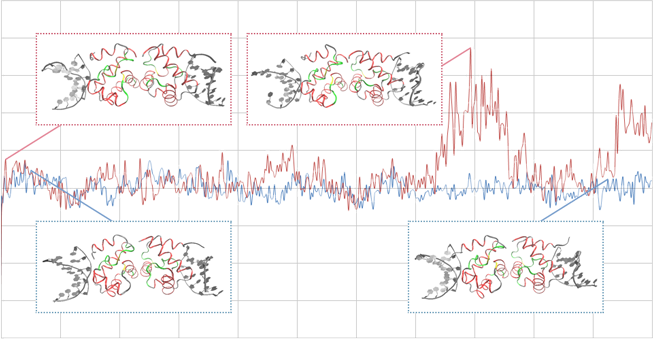
This figure shows the stability of BAF as simulated by a molecular dynamics analysis.
The blue line indicates the change in the energy of wild-type BAF with the time progress and the red line shows that for mutant BAF (p. A12T).
The inserted figures indicate the snapshots of the three-dimensional structures of wild-type and mutant BAF, respectively.
Using such a simulation analysis, we believe that we can investigate the change in the tertiary structure of BAF induced by the mutation causing NGPS.

Hutchinson-Gilford progeria syndrome (HGPS)
One of the laminopathies is Hutchinson-Gilford progeria syndrome (HGPS), a progeria syndrome, which is caused by the synthesis of an abnormal protein called progerin. Progerin is produced due to the mutation of the LMNA gene encoding Lamin A, a main component of the nuclear lamina.
The nuclear lamina provides a framework for the nuclear envelope and plays various roles in nuclear functions. Network disturbances are caused by the mutations of the genes encoding the proteins located at the nuclear lamina, which is thought to lead to the onsets of progeria syndromes.
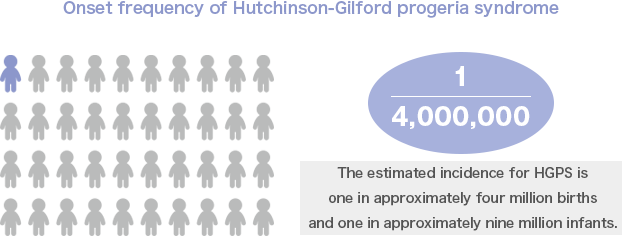
HGPS is an extremely rare disease with an estimated incidence of one in approximately four million births. However, we believe that this disease has a relatively high degree of recognition because a documentary about a patient with HGPS was broadcast in the Japanese media previously.
HGPS patients show symptoms such as a growth retardation, skin aging, hypercholesterolemia, or arteriosclerosis from infancy.
HGPS progresses rapidly and the average life span of a patient is approximately 13 years. The patient dies mainly due to cardiac functional disorders, such as myocardial infarction. To date, some research results for the treatment of HGPS at the cultured cell level have been obtained from experiments in which some chemical compounds are effective in suppressing production of the disease-causing protein, called progerin. However, the core therapeutics for this disease have not been developed yet.
Emery-Dreifuss muscular dystrophy (EDMD)

One of the laminopathies is Emery-Dreifuss muscular dystrophy (EDMD), which is a rare type of muscular dystrophy and its incidence is estimated as approximately one in 100,000 people. EDMD is one of the laminopathies that are caused by the mutation of the EMD gene encoding Emerin. Emerin is located to the nuclear inner membrane by interacting with the nuclear lamina.
Emerin has been shown to have various functions such as cell proliferation, regulation of cytoskeletons, and formation of nuclear membranes.
In addition to EMD, the LMNA and FHL1 genes, which encode Lamin A, i.e., a major component protein of the nuclear lamina and an ion-channel-binding protein, respectively, were found to be causative genes of EDMD. EDMD develops in the childhood period and is characterized by three major symptoms, such as elbow contractures, posterior region of neck, and Achilles tendon, progressive amyotrophy and muscle weakness of upper arms and lower legs, and cardiomyopathy with cardiac conduction disorders.
Patients are subjected to symptomatic treatment by orthopedic surgery; however, the prophylaxis and therapeutics for EDMD have not yet been developed because the underlying mechanism for the disease has not been elucidated yet.
Our research subject

Our research goals are to elucidate the underlying mechanisms for laminopathies, including rare and intractable diseases, and develop new therapies for these diseases using methods from various research fields, including proteomics, which is a comprehensive analysis of proteins, in silico protein structural analysis, and drug design.
We will initially obtain knowledge for the elucidation of the underlying mechanisms for these three laminopathies by investigating the disturbance of the protein-protein network induced by the mutations of causative genes.
We will then elucidate the disturbance of the protein-protein network at a molecular level using in silico protein structural analysis and perform a molecular design of a candidate compound evoking a correction of the disturbance. Finally, we will search for a therapeutic drug candidate for the disease and validate its efficacy through experiments using model cultured cells of the diseases.
Project members
Toshiya HayanoProfessor, College of Life Sciences, Department of Biomedical Sciences
Takeshi KikuchiSpecially-Appointed Professor, College of Life Sciences
Taro MannenAssistant professor, College of Life Sciences, Department of Biomedical Sciences
Other Research subjects
Cause elucidation of transporter dysfunction in Fanconi syndrome and Bartter syndrome.
Fanconi syndrome (FS) is the general functional disorder of proximal tubules, and is a syndrome in which many substances like glucose, amino acids and phosphate cannot be reabsorbed and wasted in urine. Consequently, rickets is observed in child patients whereas osteomalacia is observed in adult patients. In this research program, we try to study the detailed development mechanism and preventation of Fanconi syndrome (FS).
In this research program, we try to study the detailed development mechanisms of Fanconi syndrome (FS) and Bartter syndrom (BS), and their preventation.
Functional analysis of causative genes for Rett syndrome
Rett syndrome (RTT) is a rare disease that is associated with mental retardation, epilepsy, autism, stereotyped movements of the hands. RTT affects 1 in 10,000~15,000 girls worldwide, and it is reported that an estimated 5,000 patients exist in Japan.
In this research, we aim to propose therapeutic options by clarifying the mechanism of the onset of Rett syndrome.
Functional analysis of the gene products related to Prader-Willi syndrome
Prader-Willi syndrome (PWS) is a neurodevelopmental disorder caused by loss of expression of paternal genes on chromosome 15. Major characteristics includes short stature, obesity, hypogonadism, respiratory distress, pain insensitivity and cognitive delays.
We will focus on the features of PWS that are related to obesity in order to understand the mechanisms involved in its pathogenesis and to explore novel therapeutic options.
