
本研究の目的
レット症候群の原因遺伝子として、メチル化DNA結合タンパク質のMeCP2、タンパク質リン酸化酵素のCDKL5および転写因子であるFOXG1が知られていますが、これら遺伝子の変異とレット症候群の病態との関係はほとんど分かっていません。
最近我々は非典型的レット症候群の患者さんより新規のCDKL5遺伝子変異 (Y177C) を見出しました。そこで、本研究ではCDKL5の遺伝子変異を中心に、レット症候群の発症メカニズムを明らかにし、最終的に治療薬の創製に結び付けたいと考えています。
レット症候群とは

レット症候群は、神経系を主体とした特異な発達障害で、精神遅滞、自閉症、手もみ運動の常同運動等の症状を有する稀な疾患です。この病気の原因遺伝子としては、MeCP2、CDKL5、FOXG1等が見いだされています。
 レット症候群の詳細について
レット症候群の詳細について
症 状

MeCP2遺伝子の変異によって引き起こされるレット症候群の症状としては、乳児期より見られる自閉傾向、知的障害が挙げられます。また、手をたたいたり、もんだりといった動作を繰り返すことがこの疾患の大きな特徴とされています。また、てんかん発作、けいれん、呼吸の異常などを引き起こすことも多く見られます。
CDKL5遺伝子やFOXG1遺伝子の変異によって引き起こされる疾患は非典型的レット症候群と命名されています。CDKL5遺伝子の変異を持つ患者さんはMeCP2変異を持つ患者さんと比べ、早期にてんかん発作を引き起こすことが知られており、FOXG1遺伝子変異を持つ患者さんは小頭症を発症します。
病 因
上記の通り、MeCP2、CDKL5、FOXG1の遺伝子変異による機能喪失がレット症候群の病因となることが明らかにされています。
治 療
現在までに効果的な治療、予防法は確立されておらず、対症療法のみが行われています。
本研究の課題
レット症候群の原因遺伝子として、メチル化DNAに結合し、エピゲノム制御に関連するタンパク質であるMeCP2が同定されています。レット症候群患者さんの多くはこのMeCP2の変異を有することが明らかにされていますが、一部の患者さんはCDKL5やFOXG1といった遺伝子の変異を有し、それにより非典型のレット症候群を発症することが知られています。
私どもは、この非典型例のレット症候群の日本人患者さんからCDKL5の新規遺伝子変異 (Y177C) を見いだしています。この新規変異部位はCDKL5の触媒部位に存在していたため、この変異によってCDKL5の触媒活性が消失していると考えられました。
そこで、正常型と変異型のCDKL5精製酵素を用いて、試験管内でその酵素活性を測定したところ予想通り活性を検出できませんでした。この知見は、この分野では世界で初めて遺伝子変異とその酵素活性消失を結びつけた報告となりました。
しかしながら、CDKL5の活性消失とレット症候群の病態との関りは全くもって明らかにされていないため、私どもはCDKL5の変異が病態におよぼすメカニズムについて分子レベル、細胞レベルで明らかにすべく、研究に取り組んでいます。
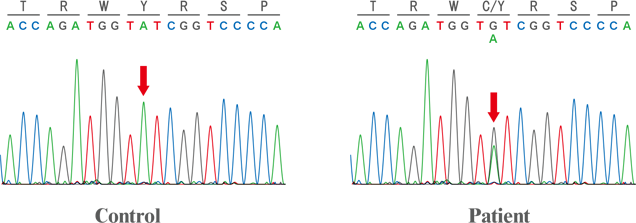
レット症候群患者さんのCDKL5遺伝子配列を解析し、CDKL5の新規の変異部位を決定しました (上図赤矢印部)。この変異によりCDKL5の177番目のチロシン(Y)がシステイン(C)へと変化することが明らかになりました。
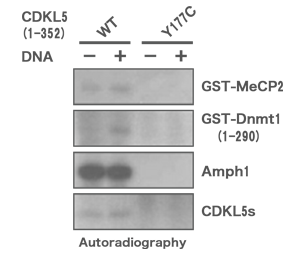
上記の変異CDKL5の酵素活性について検討したところ、CDKL5 (Y177C) は既知のCDKL5の基質タンパク質をリン酸化することができませんでした。
プロジェクトメンバー
薬学部・薬学科・教授稲津 哲也
Tetsuya Inazu
生命科学部・生命情報学科・教授伊藤 將弘
Masahiro Ito
薬学部・薬学科・准教授河野 貴子
Takako Kawano
生命科学部・生命情報学科・助教久保田 幸彦
Yukihiko Kubota
その他の研究テーマ
ファンコニ症候群におけるトランスポーターの機能不全の原因解明
ファンコニ症候群は、近位尿細管における膜輸送全般が障害を受けることでブドウ糖、アミノ酸やリンなどの再吸収全般が障害されておこる発育不全や骨軟化症などを特徴とします。
本研究では、ファンコニ症候群の発症機構を解明し、予防・治療法について検討していきます。
プラダー・ウィリー症候群の原因タンパク質の機能解明
プラダー・ウィリー症候群は、15番染体の同症候群責任領域の欠損によって発症する神経発達障害疾患です。臨床症状としては、低身長、肥満、性腺機能障害、呼吸異常、末梢感覚障害、精神発達遅延など様々です。
本研究では、プラダー・ウィリー症候群の肥満の症状に焦点をあてて、そのメカニズムを明らかにすることで治療法の提案を行いたいと考えています。
核膜病の発症機構の解明と治療法の開発
本研究では、プロテオミクス、細胞生物学、in silicoタンパク質構造解析および医薬分子設計の最先端の研究手法に基づいて、2つの核膜病を標的とし、発症機構を領域横断的に解析することで、新たな治療法を開発することを目的としています。
