
本研究の目的
本研究では、3つの核膜病を標的とし、プロテオミクス、細胞生物学、in silico (コンピュータを用いた) タンパク質構造解析および医薬分子設計の最先端の研究手法に基づいて、それぞれの疾患原因タンパク質の網羅的な相互作用タンパク質解析を行うことで、疾患の発症機構に関わるタンパク質間ネットワークを特定し、新たな治療法を開発することを目的としています。
核膜病とは

細胞核の内部にはゲノムDNAが収納されており、そこに刻まれた遺伝情報の発現が核内に局在する様々なタンパク質が関わる精緻な機構によって制御されています。
核は、二重膜によって細胞質と隔たれており、その内側にはラミンAを主要な構成成分とする核ラミナという裏打ち構造があります。
核ラミナには多くのタンパク質が局在し、多様な核機能の制御に重要な役割を担っています。これらの核膜に局在するタンパク質をコードする遺伝子の突然変異に伴い発症する病気は「核膜病」と呼ばれています。
本研究では、「核膜病」に分類される稀少疾患であるNéstor-Guillermo Progeria Syndrome (ネスター・ギラーモ早老症:NGPS)、Hutchinson-Gilford Progeria Syndrome (ハッチンソン・ギルフォード早老症:HGPS)、およびEmery-Dreifuss Muscular Distrophy (エメリー・ドレフィス型筋ジストロフィー:EDMD) の発症機構の解明とこれらの病気の治療法の開発を目指します。
 ネスター・ギラーモ早老症:NGPS
ネスター・ギラーモ早老症:NGPS
早老症のひとつであるネスター・ギラーモ早老症 (NGPS) は、核ラミナに結合するタンパク質と相互作用することで核内膜に局在するタンパク質であるBarrier-to-autointegration factor (BAF) の遺伝子 (BANF1) の突然変異によって発症する核膜病のひとつです。
BAFは、様々な遺伝子の発現や紫外線などにより損傷を受けたDNAの修復過程において重要な役割を果たしていると報告されていることから、NGPSの発症原因である変異により、BAFのDNAとの結合能が低下することでこれらのBAFの機能が低下し、早老症が発症すると考えられています。

NGPSは、およそ100万人に1人の頻度で発症し、幼少期から成長障害、皮膚の乾燥・委縮、骨粗鬆症および骨融解などの症状を呈します。一方で、成人期の初期には、糖尿病、高トリグリセリド血症、心血管障害などの兆候は見られず、患者さんの早老症の進行は、HGPSなどに比べて緩やかに進行することから患者さんの平均寿命も長いため、NGPSは慢性早老症と定義されています。
これまでに、NGPSの発症機構の詳細は明らかにされていないことから、その予防法および治療法は開発されていないのが現状です。
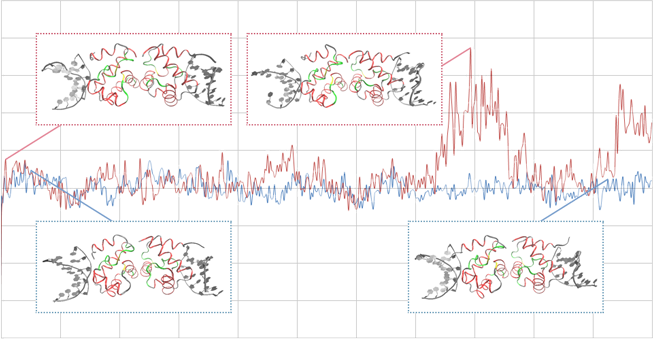
この図は、NGPS原因タンパク質BAFの分子動力学シミュレーションの結果の一部を示しています。
青線は正常なBAFのエネルギーの時間的変化、赤線はBAFの変異体 (12番目の アラニンがスレオニンに変異したもの) の変化を示しています。
挿入されている図はそれぞれのスナップショットのタンパク質の立体構造の図です。
このようなシミュレーション解析から、NGPS発症の原因となる変異に伴うBAFタンパク質の立体構造の変化を調べることができると考えられます。

ハッチンソン・ギルフォード早老症:HGPS
早老症のひとつであるハッチンソン・ギルフォード早老症 (HGPS) は、核ラミナの主要構成成分であるラミンAの遺伝子 (LMNA) の突然変異により、プロジェリンと呼ばれる異常なタンパク質が産生されることで発症する核膜病のひとつです。
核ラミナは、様々な核機能を司る重要な核膜裏打ち構造であり、その構成タンパク質の変異によりこれらの核機能の撹乱がもたらされ、早老症が発症すると考えられています。
HGPSは、およそ400万人に1人の頻度で発症する極めて稀な疾患ですが、以前、日本のメディアでHGPS患者さんのドキュメンタリーが放映されたことから、比較的認知度が高い疾患ではないでしょうか?
HGPS患者さんには、幼児期から成長遅延、皮膚の老化、高コレステロール血症、動脈硬化などの症状が現れます。
早老症の進行は早く、患者さんの平均寿命は約13歳であり、その死因は主に心筋梗塞などの心機能障害です。これまでに、LMNAの変異に伴うプロジェリンの産生を抑制する化合物を用いることで、培養細胞レベルでのHGPSの治療に向けた研究成果が得られていますが、根本的な治療法の確立には至っていません。
エメリー・ドレフィス型筋ジストロフィー:EDMD

エメリー・ドレフィス型筋ジストロフィー (EDMD) は、筋ジストロフィーの中でも稀な病型に位置付けられており、その発症頻度は約10万人に1人と報告されています。EDMDは、核ラミナと相互作用することで核内膜に局在するタンパク質であるエメリンの遺伝子 (EMD) の突然変異によって発症する核膜病のひとつです。
エメリンは、細胞増殖、細胞骨格制御、および核膜構築などの機能をもつことが示されています。
EDMDの原因遺伝子としてEMDが同定されて以降、核ラミナの主要構成タンパク質ラミンAおよびイオンチャネル結合タンパク質FHL1をコードする遺伝子が病因遺伝子として報告されています。EDMDは、幼少期に発症し、肘部、後頸部、アキレス腱などの拘縮と進行性の上腕下腿型の筋委縮や筋力低下、心伝導障害を伴う心筋症という三大症状を特徴とする進行性筋ジストロフィーです。
整形外科手術による対処的な治療が施されているものの、EDMDの発症機構の詳細は不明であることから、その予防法および根本的な治療法は開発されていないのが現状です。
本研究の課題

本研究では、タンパク質の網羅的な解析法であるプロテオミクス、in silico (コンピュータを用いた) タンパク質構造解析、および医薬分子設計といった異なった研究分野を融合させることで、稀少疾患・難治性疾患である核膜病の発症機構の解明および新たな治療法の開発を目指します。
まず、3つの核膜病について、それぞれの疾患原因タンパク質の変異に伴って、どのようなタンパク質間相互作用に乱れが生じるのかを明らかにすることで疾患の発症機構解明につながる知見を得ます。
次に、in silicoタンパク質立体構造解析により、疾患発症の原因である変異に伴うタンパク質間ネットワークの撹乱を分子レベルで解明し、撹乱の修正を誘起する候補化合物の分子設計を行います。さらに、疾患モデル培養細胞を用いて、当該疾患治療薬の候補化合物の探索とその有効性の検証を進めます。
プロジェクトメンバー
生命科学部・生命医科学科・教授早野 俊哉
Toshiya Hayano
生命科学部・特任教授菊地 武司
Takeshi Kikuchi
生命科学部・生命医科学科・助教萬年 太郎
Taro Mannen
その他の研究テーマ
ファンコニ症候群におけるトランスポーターの機能不全の原因解明
ファンコニ症候群は、近位尿細管における膜輸送全般が障害を受けることでブドウ糖、アミノ酸やリンなどの再吸収全般が障害されておこる発育不全や骨軟化症などを特徴とします。本研究では、ファンコニ症候群の発症機構を解明し、予防・治療法について検討していきます。
レット症候群における原因遺伝子の機能解明
レット症候群は、神経系を主体とした特異な発達障害で、精神遅滞、てんかん、自閉症、常同運動等 (手もみ運動等) の症状を有し、頻度としては女児の10,000人~15,000人に1人の割合で発症、日本では推定5,000人の患者さんが存在すると報告されている稀少疾患です。
本研究では、レット症候群の発症メカニズムを明らかにすることで、治療法の候補の提案を行いたいと考えています。
プラダー・ウィリー症候群の原因タンパク質の機能解明
プラダー・ウィリー症候群は、15番染体の同症候群責任領域の欠損によって発症する神経発達障害疾患です。臨床症状としては、低身長、肥満、性腺機能障害、呼吸異常、末梢感覚障害、精神発達遅延など様々です。
本研究では、プラダー・ウィリー症候群の肥満の症状に焦点をあてて、そのメカニズムを明らかにすることで治療法の提案を行いたいと考えています。
