
本研究の目的
プラダー・ウィリー症候群 (以下PWS) は、15番染色体のPWS責任領域の欠損による神経発達障害疾患です。PWS責任領域上にはさまざまな遺伝子が存在しますが、その中でNecdin (遺伝子はNDNと表記) タンパク質に焦点を当てて発症機構を解明し、治療法について検討していきます。
プラダー・ウィリー症候群とは
PWSは、突発的な15番染色体のPWS責任領域の欠損による神経発達障害疾患で、さまざまな神経症状を呈します (詳細の症状参照)。図1はPWS責任領域の遺伝子マップです。
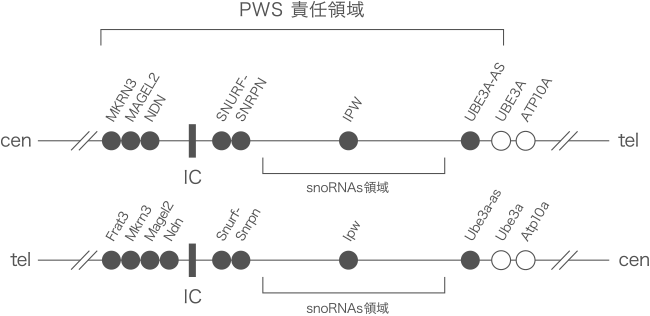
(図1) PWS責任領域の遺伝子マップ
上段はヒト15番染色体 (15q11-q13)、下段はマウス7番染色体 (7B/C) のPWS責任領域周辺の遺伝子を示しています。
黒丸は父親由来の染色体のみから発現する遺伝子を現しています。snoRNAsの領域には多数の非翻訳RNA遺伝子を含んでいます (IPWはsnoRNA遺伝子ではない)。
ICはインプリンティングセンターで、この領域全体のインプリンティングを調節しています。
上段はヒト15番染色体 (15q11-q13)、下段は上段の対応するマウス7番染色体 (7B/C) のマップです。PWS責任領域は特殊な領域で、ゲノムインプリンティングと呼ばれるDNA修飾によって父親由来の染色体のみから遺伝子発現が起こります。このインプリンティングは、図で示すIC (インプリンティングセンター) の作用によって起こります。多くの場合、PWSではこの責任領域上の全ての遺伝子発現が欠損することによって、中枢・末梢神経系の発達障害が起こりさまざまな症状が起こります。しかし、これら責任領域上のどの遺伝子がどういう作用で、その欠損がどういう症状に関連しているかよくわかっていません。
 プラダー・ウィリー症候群の詳細について
プラダー・ウィリー症候群の詳細について
原因
PWSは、1/15,000-1/30,000の頻度で突発的に起こる染色体異常が原因です。染色体異常のパターンは以下の図2の通りです。

(図2) PWSの原因
Pは父親由来染色体で、Mは母親由来染色体を指します。父親由来15番染色体のPWS責任領域欠損が65-75%と最も多くなります。ユニペアレンタルダイソミーは、2本の15番染色体とも母親由来である場合です。したがってどちらの染色体からも発現しません。
Pは父親由来染色体で、Mは母親由来染色体を指します。
最も多いのは欠損タイプで、PWS責任領域上の遺伝子は父親由来染色体のみから発現するため、父親由来染色体上のPWS責任領域の欠損によるものです。次に多いのはユニペアレンタルダイソミーで、15番染色体が2本とも母親由来染色体となっているものです。
この場合はどちらの染色体からもPWS責任領域上の遺伝子は発現しないことになります。
インプリンティングエラー頻度は少ないですが、父親由来染色体上のPWS責任領域もメチル化によって遺伝子発現しなくなるものです。
診断は、染色体検査によってDNAのメチル化や欠損の有無によって行われます。
特に、PWS責任領域上のSNRPN遺伝子の欠損の有無、プロモーター領域のメチル化の有無によって行われます。
症状
PWSのおもな症状
- 筋緊張低下とそれに伴う運動の低下、
母乳などの摂食不良 - 視床下部の異常による性腺発達障害、
外性器発達障害、二次性徴の遅れ - 精神発達遅延
- 低身長
- 1-4才位より始まる過食とそれに伴う肥満
(満腹感が得られない) - 行動異常
(かんしゃく気質、頑固、強迫症で同じことを繰り返す)
PWSのその他の症状
- 睡眠時無呼吸
- 手足が小さい
- 斜視
- てんかん
- 骨軟化症/骨粗鬆症
- 唾液量の低下
- 痛み域値の上昇 (痛みを感じ難い)
- 皮膚を引っ掻く
治療
PWSに対する治療は、対処療法が中心となります。
- 乳児期における適切な栄養の確保
- 成長ホルモンの補充
- 性ホルモンの補充
- 強迫症に対してセロトニンアゴニスト服用
- 職業訓練、言語療法など

本研究の課題
PWS責任領域上にあるNecdin遺伝子に焦点を当てて解析を行っていきます。
これまでにNecdin遺伝子欠損マウスが、視床下部ホルモン産生ニューロンの減少、睡眠時無呼吸発作、感覚ニューロンの細胞死増大による痛み域値の上昇、皮膚を引っ掻く行動などのPWS類似の表現型を示すことからPWSの症状の一旦を担うと考えられます。
Necdin遺伝子欠損マウスの特に視床下部ニューロンの発達異常や機能異常について解析を行い、作用メカニズムを明らかにします。
Necdinはヒトとマウスでは30種類以上存在するMAGEファミリータンパク質の一つですが、ニワトリより下等な生物では単一のMAGEタンパク質であるNse3が発現しています。
したがってモデル生物である線虫と細胞性粘菌を利用することでプロトタイプの機能解析が可能となります。そこで、これらのモデル生物を用いてNse3とその結合タンパク質であるNse1とNse4の発現と局在を解析し、低発現線虫や遺伝子改変粘菌を作成し、これら遺伝子の機能を解明していきます。
in silicoの手法による立体構造解析の結果にもとづいて、Necdinやそのシグナル伝達タンパク質と相互作用する化合物をデザインします。
化合物ライブラリーから類縁物質を選択し、それぞれの効果をin vitroの培養細胞再構成系で確認するとともに、作用物質についてはNecdin遺伝子欠損マウスに投与し、摂食行動と高脂肪食での肥満誘発について解析し治療効果を確認します。また、遺伝子欠損マウスの視床下部に別のグループが開発したラットiPS由来の神経細胞を導入し、症状の改善効果の解析を行っていきます。
プロジェクトメンバー
薬学部・薬学科・教授谷浦秀夫
Hideo Taniura
生命科学部・生命医科学科・教授田中秀和
Hidekazu Tanaka
薬学部・創薬科学科・教授鈴木 健二
Kenji Suzuki
薬学部・薬学科・助教正木 聡
So Masaki
薬学部・助教添田修平
Shuhei Soeda
生命科学部・講師中谷仁
Jin Nakatani
生命科学部・生命医科学科・助教澤野俊憲
Toshinori Sawano
その他の研究テーマ
ファンコニ症候群におけるトランスポーターの機能不全の原因解明
ファンコニ症候群は、近位尿細管における膜輸送全般が障害を受けることでブドウ糖、アミノ酸やリンなどの再吸収全般が障害されておこる発育不全や骨軟化症などを特徴とします。
本研究では、ファンコニ症候群の発症機構を解明し、予防・治療法について検討していきます。
レット症候群における原因遺伝子の機能解明
レット症候群は、神経系を主体とした特異な発達障害で、精神遅滞、てんかん、自閉症、常同運動等 (手もみ運動等) の症状を有し、頻度としては女児の10,000人~15,000人に1人の割合で発症、日本では推定5,000人の患者さんが存在すると報告されている稀少疾患です。
本研究では、レット症候群の発症メカニズムを明らかにすることで、治療法の候補の提案を行いたいと考えています。
核膜病の発症機構の解明と治療法の開発
本研究では、プロテオミクス、細胞生物学、in silicoタンパク質構造解析および医薬分子設計の最先端の研究手法に基づいて、2つの核膜病を標的とし、発症機構を領域横断的に解析することで、新たな治療法を開発することを目的としています。
